 ESPCI web site
ESPCI web site


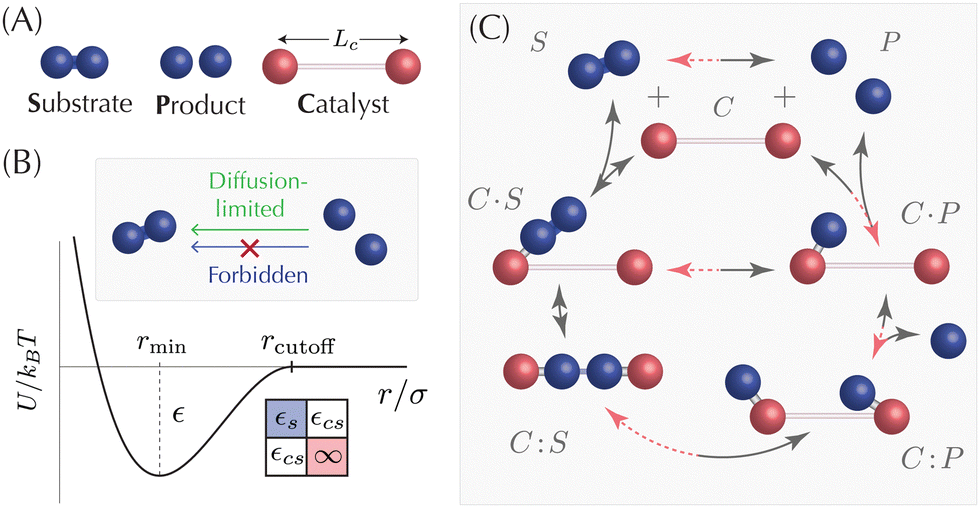
Catalysis, the acceleration of chemical reactions by molecules that are not consumed in the process, is essential to living organisms but remains absent in physical systems that aspire to emulate biological functionalities with artificial components. Here we demonstrate how to design a catalyst using spherical building blocks interacting via programmable potentials, and show that a minimal catalyst design, a rigid dimer, can accelerate a ubiquitous elementary reaction, the cleaving of a bond. Combining coarse-grained molecular dynamics simulations and theory, and by comparing the mean reaction time for bond dissociation in the presence and absence of the catalyst, we derive geometrical and physical constraints for its design and determine the reaction conditions under which catalysis emerges in the system. The framework and design rules that we introduce are general and can be applied to experimental systems on a wide range of scales, from micron size DNA-coated colloids to magnetic handshake materials in the macroscale, opening the door to the realization of self-regulated artificial systems with bio-inspired functionalities.
ROYAL SOCIETY OF CHEMISTRY
By: Maitane Muñoz-Basagoiti, Olivier Rivoire, Zorana Zeravcic.
First published on 15th May 2023
DOI: https://pubs.rsc.org/en/content/art...